Research
A Drosophila model of Charcot-Marie-Tooth neuropathy
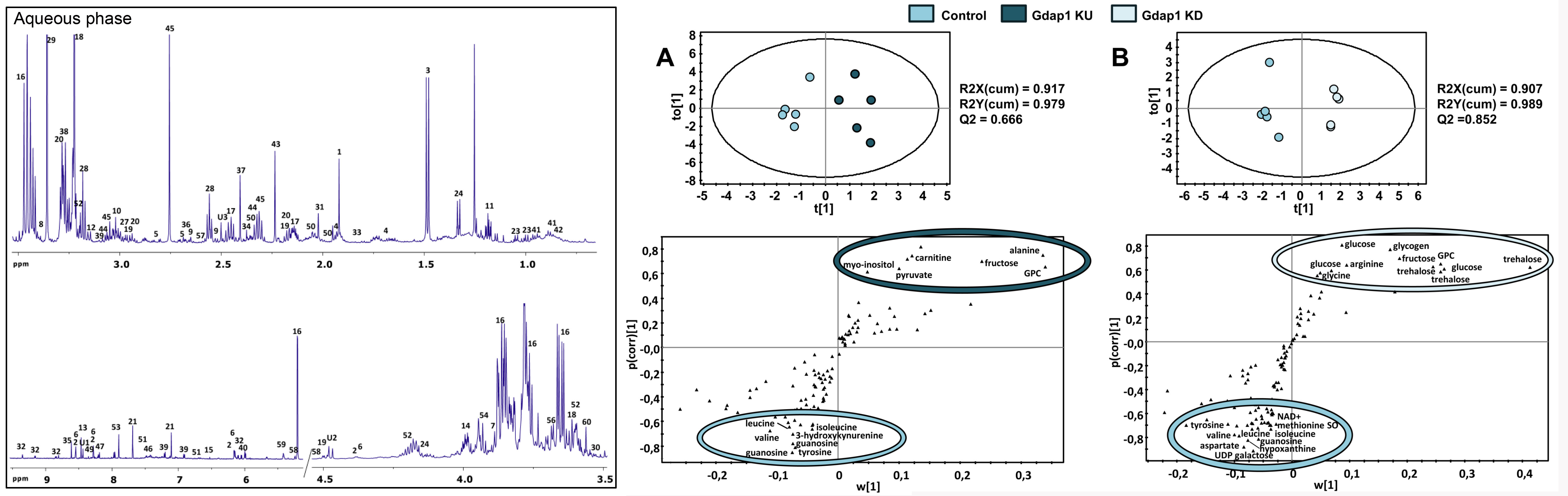
We generated a model of Charcot-Marie-Tooth neuropathy caused by mutations in the GDAP1 gene. We demonstrated that this gene has an ortholog in Drosophila, which we named Gdap1, and by altering its expression levels we could study the neuromuscular degeneration. We found that some candidate disease mechanisms such as oxidative stress only occur late, and discovered an unexpected alteration in insulin signaling and energy metabolism. To uncover metabolic alteration in these models, and candidate biomarkers for clinical translation, we performed a metabolic analysis by unbiased nuclear magnetic resonance metabolomics followed by transcriptomic analysis of those pathways in which we detected alterations. Although the metabolome of young flies was largely unaltered, in old flies we could see clear changes in energy metabolism: accumulation of carbohydrates and reduction of lipids. We saw consistent changes in the transcript levels of carbohydrate and lipid metabolism, likely due to an inactivation of the pyruvate dehydrogenase complex. Although these changes were only evident in aged flies, we found alterations in insulin signaling already in young flies: increased levels of expression of the genes coding for the Drosophila insulin-like peptides (Dilps) and also of markers of IIS down-regulation: the insulin receptor (InR) and 4EBP. In addition, both models had a reduced ratio of phosphorylated to total Akt protein. A series of experiments in collaboration with Prof. Axel Methner (Johannes Gutenberg University of Mainz), who already had a system of over-expression of GDAP1 mutations in a human neuroblastoma cell line, also showed alterations consistent with a down-regulation of insulin signaling: translocation of FOXO to the nucleus, and a reduced ratio of phosphorylated to total protein for AKT and its target GSK3.
We have also studied the Drosophila junctophilin gene, equivalent to the four human JPH1-4 genes, whose mutations have important implications in pathology as modifiers of GDAP1 (JPH1), or causative of genetic myocardiopathy (JPH2) and Huntinton-disease-like 2 (JPH3). We have demonstrated that this gene is a modifier of pathological expansions in the Huntingtin gene and of the Notch signaling pathway, which can shed light in the disease mechanisms of the mutations in the human Junctophilins.
Personalized medicine in Dravet Syndrome
Dravet syndrome is a childhood epilepsy, babies are born healthy, but they soon begin to manifest periodic epileptic seizures. In addition to the inherent danger of an epileptic seizure, with age these can cause a cognitive and behavioral deterioration, and can even cause the death of the infant. Dravet syndrome is a disease with very low prevalence, currently estimated at 1 infant every 20,000 to 40,000 births. In 80% of cases, this pathology is caused by the mutation in one of the alleles of the SCN1A gene, which codes for the α-subunit of the voltage-dependent sodium channel type I. Although most patients have mutations in the SCN1A gene, and these are always dominant, each mutation produces a different clinical picture and responds in different ways to anticonvulsant treatments. Hence the need to use personalized medicine to seek new treatments, as there probably will not be a single drug that can treat all forms of the disease. At this time there are no curative treatments for this syndrome, only palliative. Drosophila melanogaster has a gene homologous to the human SCN1A gene, and mutations in this gene are associated with epileptic episodes similar to those that occur in patients.
In collaboration with the patient association ApoyoDravet we are developing a project in which we will reproduce the mutations of Spanish and Ibero-American patients in Drosophila through genomic editing. We use a technology specific to our model by which we inactivate the Drosophila gene and instead introduce a human cDNA in which we have reproduced the clinical mutations. We hope that these precision models allow us to know the clinical differences between patients and help us find personalized treatments.

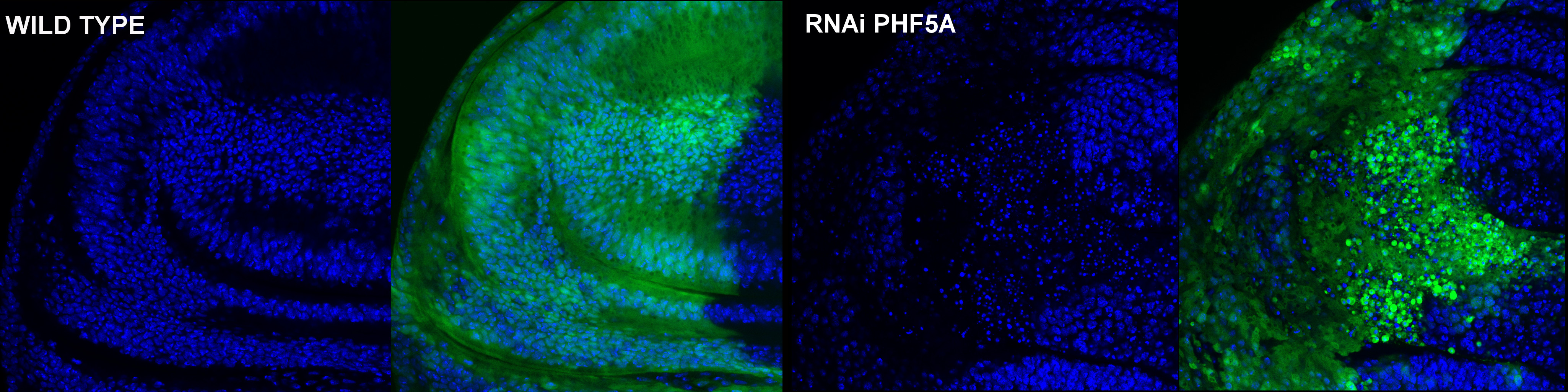
Novel functions for the PHF5A protein
The PHF5A protein is highly conserved from unicellular eukaryotic organisms, such as yeast, to humans. Several studies have demonstrated its involvement in splicing, in transcriptional processes associated with the response to estrogen and in growth and development of Schizosaccharomyces pombe and Caenorhabditis elegans. In addition, it has been cataloged as a possible therapeutically target gene for glioblastoma. We have generated stable transgenic Drosophila melanogaster strains with adjustable levels of PHF5A for the study of protein expression and function. Our results are the first study of PHF5A in a whole animal model, and suggest a completely novel function for this protein. Its expression is ubiquitous and localises mainly to the nucleus. This expression and localization pattern changes during the different phases of mitosis. PHF5A is essential for D. melanogaster development, since individuals with limiting amounts of PHF5A cannot progress beyond the third larval instar and the number of mitoses is compromised in proliferative tissues. PHF5A silencing blocks the cells in G2/M too a fact that induces a mitotic catastrophe. In contrast, Phf5a overexpression does not produce any evident phenotype. The evidence points toward a requirement for chromatin cohesion during mitosis.