Investigación
Un modelo en Drosophila de la neuropatía de Charcot-Marie-Tooth
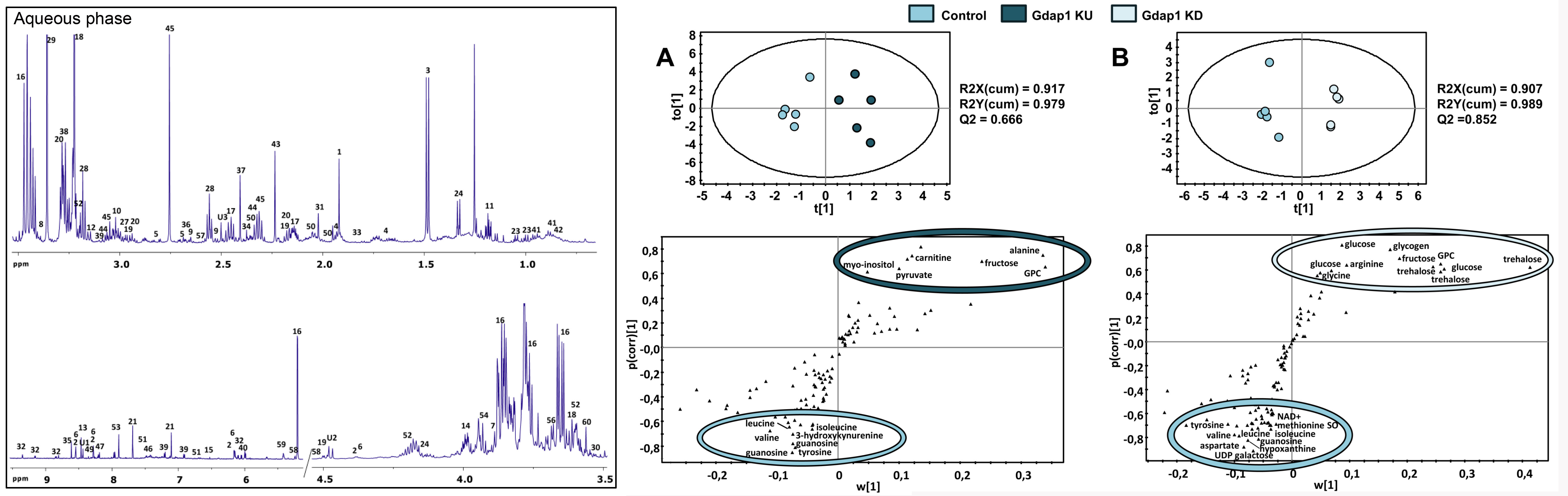
En nuestro grupo generamos un modelo de neuropatía de Charcot-Marie-Tooth causada por mutaciones en el gen GDAP1. Demostramos que este gen tiene un ortólogo en Drosophila, que llamamos Gdap1, y demostramos que la alteración de sus niveles de expresión causa degeneración neuromuscular. Algunos mecanismos de enfermedad candidatos, como el estrés oxidativo, solo aparecen tarde y hemos descubierto una alteración inesperada en la señalización por y el metabolismo energético. Para descubrir la alteración metabólica en estos modelos, y definir biomarcadores candidatos para su traslación clínica, realizamos un análisis metabólico no sesgado mediante metabolómica de resonancia magnética nuclear, seguido de análisis transcriptómico de las vías en las que detectamos alteraciones. Aunque el metaboloma de las moscas jóvenes no se modificó en gran medida, en moscas viejas pudimos ver cambios claros en el metabolismo energético: acumulación de carbohidratos y reducción de lípidos. Vimos cambios consistentes en los niveles de transcripción del metabolismo de carbohidratos y lípidos, probablemente debido a una inactivación del complejo piruvato deshidrogenasa. Aunque estos cambios solo fueron evidentes en las moscas viejas, encontramos alteraciones en señalización por insulina ya en las moscas jóvenes: aumento de los niveles de expresión de los genes que codifican los péptidos de Drosophila similares a la insulina (Dilps) y también de los marcadores de inhibición de la ruta: el receptor de la insulina (InR) y 4EBP. Además, ambos modelos tenían una relación reducida de proteína Akt fosforilada a total. Una serie de experimentos en colaboración con el Prof. Axel Methner (Universitat Johannes Gutenberg de Maguncia, Alemania), que ya tenía un sistema de sobreexpresión de mutaciones de GDAP1 en una línea celular de neuroblastoma humano, también mostró alteraciones consistentes con una regulación negativa de señalización de insulina: translocación de FOXO al núcleo, y una relación reducida de proteína fosforilada a proteína total para AKT y su diana GSK3.
También hemos estudiado el gen junctophilin de Drosophila, equivalente a los cuatro genes humanos JPH1-4, cuyas mutaciones tienen importantes implicaciones en la patología como modificadores de GDAP1 (JPH1), o causante de miocardiopatía hereditaria (JPH2) y Huntinton-disease-like 2 (JPH3). Hemos demostrado que este gen es también un modificador de las expansiones patológicas en el gen Huntingtin y de la vía de señalización de Notch, lo cual puede arrojar luz en los mecanismos de la enfermedad de las mutaciones en los genes JPH1-4 humanos.
Medicina personalizada en Síndrome de Dravet
El síndrome de Dravet es una epilepsia infantil, los bebés nacen sanos, pero pronto empiezan a manifestar crisis epilépticas periódicas. Además del peligro inherente a una crisis epiléptica, con la edad estas pueden causar un deterioro cognitivo y conductual, y puede incluso causar la muerte del infante. El síndrome de Dravet es una enfermedad con muy baja prevalencia, se estima que lo padece 1 infante cada 20.000 a 40.000 nacimientos. En el 80% de los casos esta patología está causada por la mutación en uno de los alelos del gen SCN1A, que codifica para la subunidad α del canal de sodio voltaje-dependiente tipo I. A pesar de que la mayoría de pacientes tienen mutaciones este único gen SCN1A, y estas son siempre dominantes, cada mutación produce un cuadro clínico distinto y responde de distinta manera a los tratamientos con anticonvulsivos. De ahí la necesidad de utilizar la medicina personalizada para buscar nuevos tratamientos, ya que probablemente no habrá un único fármaco que pueda tratar todas las formas de la enfermedad. En estos momentos no hay tratamientos curativos para este síndrome, únicamente paliativos. Drosophila melanogaster tiene un gen homólogo al gen SCN1A humano, y las mutaciones en este gen están asociadas a episodios epilépticos similares a los que se dan en los pacientes.
En colaboración con la asociación de pacientes ApoyoDravet estamos desarrollando un proyecto en que reproduciremos las mutaciones de pacientes españoles e iberoamericanos en Drosophila mediante edición genómica. Utilizamos una tecnología específica de nuestro modelo mediante la cual inactivamos el gen de Drosophila y en su lugar introducimos un cDNA humano en el que hemos reproducido las mutaciones clínicas. Esperamos que estos modelos de precisión nos permitan conocer las diferencias clínicas entre los pacientes y nos ayuden a encontrar tratamientos personalizados.

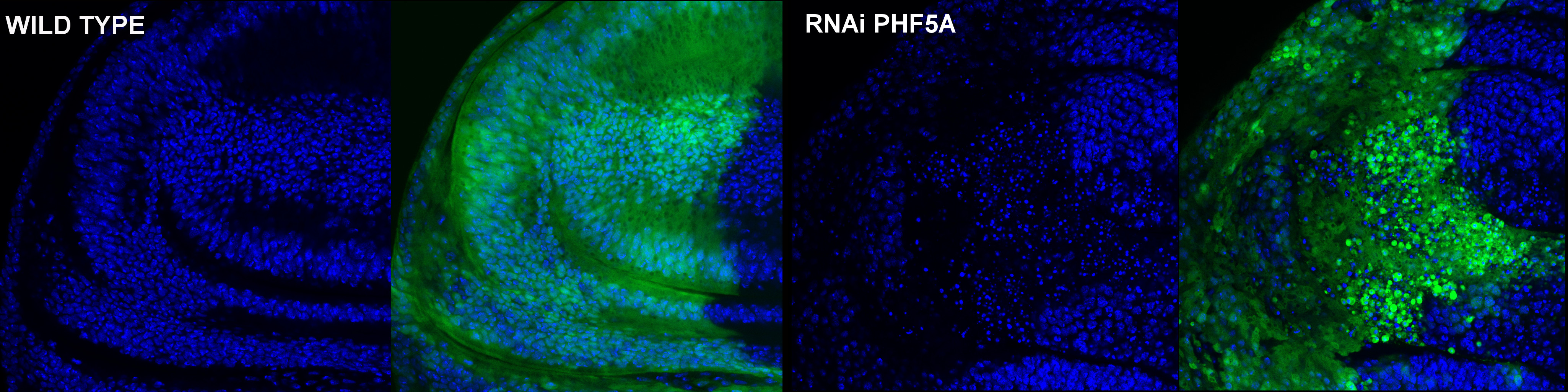
Nuevas funciones de la proteína PHF5A
La proteína PHF5A es una proteína altamente conservada desde organismos eucariotas unicelulares como las levaduras hasta el hombre. Diversos estudios demuestran su participación en el procesamiento de ARNs mensajeros, en procesos de transcripción asociados a la respuesta a estrógenos y en el crecimiento y desarrollo de Schizosaccharomyces pombe y Caenorhabditis elegans. Además, ha sido catalogada recientemente como un posible gen diana para el tratamiento del glioblastoma. Hemos generado cepas transgénicas de Drosophila melanogaster con niveles regulables de PHF5A para el estudio de la expresión y función de la proteína. Nuestros resultados representan el primer estudio de PHF5A en un modelo animal y sugieren una posible función totalmente novedosa para esta proteína. Su expresión es ubicua, con una localización presentemente nuclear. Este patrón de expresión y localización cambia durante las diferentes fases de la mitosis. El silenciamiento de PHF5A resulta letal durante el desarrollo, más concretamente en el tercer estadio larvario. El silenciamiento reduce el número de mitosis en tejidos proliferativos, debido a una parada en G2/M de las células silenciadas, induciendo una catástrofe mitótica. Por el contrario, su sobreexpresión no produce alteraciones fenotípicas detectables. La evidencia sugiere una función en la cohesión de la cromatina durante la mitosis.